Overview
Why do some cancer patients respond to treatment, while others succumb to their disease? Why are some treatments effective initially, but fail over time? How do cancer cells acquire the ability to spread from one part of the body to another? These are the fundamental and unresolved questions which motivate cancer research worldwide. Viewing cancer progression through the lens of evolutionary theory, our approach to addressing these problems centers on studying the genomes of cancer cells as fundamental units of information encoding biological properties. We have an active research program across several interrelated major topics including: Cancer evolution, single cell genomics, mutational signatures and prediction of drug response. The primary activities in the lab consist of experimental design, large-scale cancer multi-omics data analytics, development of machine learning and Bayesian statistics methods and biological study of ovarian, breast and lymphoid cancers.
Cancer Evolution
Our lab is motivated by studying cancer through the lens of evolution. We are engaged in several studies that span both temporal and spatial multi-sample studies of our cancers of interest. Observing the dynamics of genomically-defined clones reflected in timeseries biopsies of patient tumours, patient-derived xenografts, or through spreading of clones across anatomical sites is a key area of interest for our lab. For example in recent work, we have identified clonal expansion patterns underpinning breast cancer evolution, mapped the spread of clones within the peritoneal cavity of ovarian cancers and identified reproducible clonal dynamics in patient derived breast cancer xenografts. Our current questions relate to drug selection and the interplay between malignant cells and the tumour microenvironment.
We have a keen interest in learning fitness trajectories from time series study of cancer populations within controlled interventions such as CRISPR or pharmacologic methods as a means to predict response to drugs. Using extensions of population genetics theory, we are interested in predicting how cell populations will respond in the presence of a perturbation. This is indeed ‘hard’ and entails the need to decipher stochastic drift, clonal interaction and positive selection. Furthermore, drug response may not be encoded in the genome, requiring dynamic state switching through the epigenome and reflected in the transcriptome. What proportion of drug response can be explained through encoded mutations in the genome? This remains unknown. We are pursuing integrative, multimodal molecular views over time in bulk tissues and single cells as substrate to address this question.
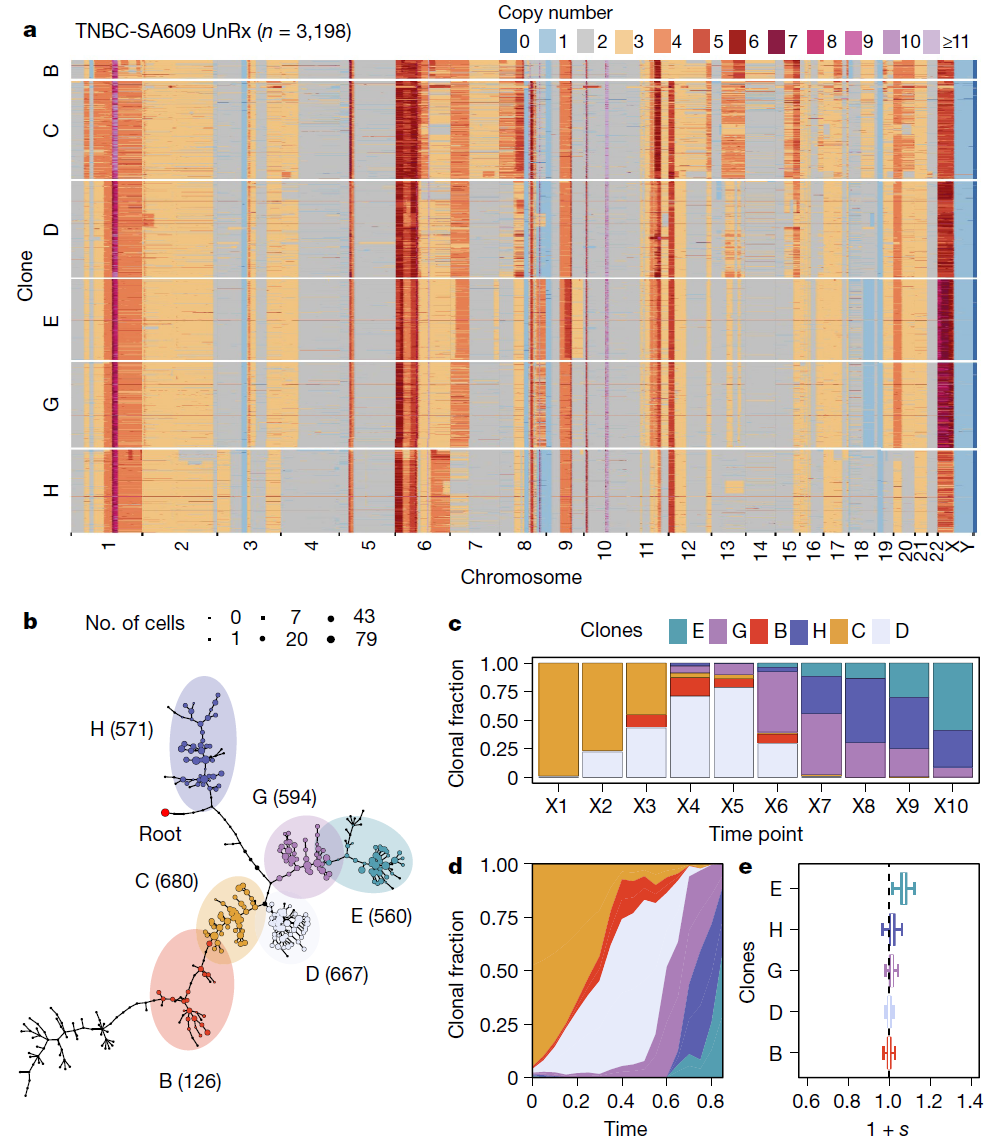
From Salehi et al. (2021, Nature), we are able to reconstruct the genomic phylogeny of an untreated triple-negative breast cancer patient derived xenograft propagated across 10 generations of mice. Using single cell DNA sequencing and statistical analysis, we are able to determine the relative fitness of cellular subpopulations and copy number events unique to each.
Relevant recent work:
- Single-cell genomic variation induced by mutational processes in cancer. Nature 612, 106-115 (2022)
- Accurate determination of CRISPR-mediated gene fitness in transplantable tumours. Nature communications 13, 4534 (2022)
- Clonal fitness inferred from time-series modelling of single-cell cancer genomes. Nature 595, 585-590 (2021)
- ddClone: joint statistical inference of clonal populations from single cell and bulk tumour sequencing data. Genome Biol 18, 44 (2017)
Single Cell Genomics of Cancer
The unit of evolutionary selection in cancer is the cell. Extraordinary progress in measurement technologies has made it possible to reliably and accurately sequence the genomes of individual cancer cells at scale. We have recently optimized biophysical techniques and hidden Markov model approaches to ascertain highly accurate copy number profiles of thousands of cancer cells. As such, studying the ‘population genetics’ of cancer cells is a tractable goal. We are developing phylogenetics and fitness computational models through measuring the population dynamics of thousands of individual cancer cells across timeseries, spatial samples and in the presence of genetic and pharmacologic intervention. These experiments and computational methods are improving our knowledge of background mutation rates, properties of positive and negative selection (not observable in bulk samples) and how phylogenetic topologies reflect the relative fitness of clones. Furthermore, as we integrate multi-modal measurements, profiling the evolving malignant population in the context of its tumour microenvironment will be a strong interest of the lab.
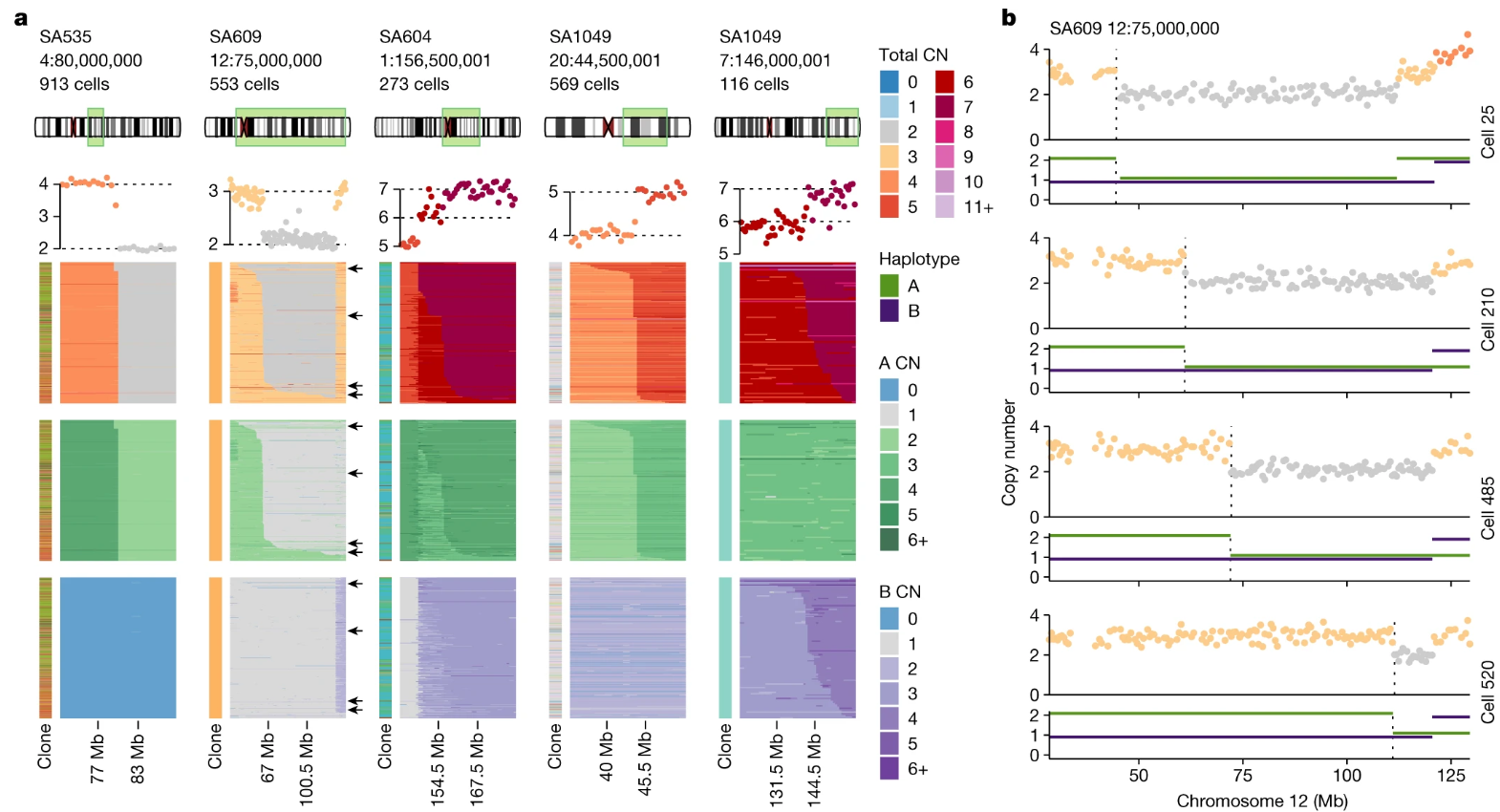
From Funnell et al. (2022, Nature), single cell whole genome sequencing has allowed us to discover novel patterns of breakpoints not detectable with bulk sequencing technologies. Here, we see “serrated” breakpoint profiles, showing an amplification event that increases across distinct different cells in the same population.
Relevant recent work:
- A Scalable Strand-Specific Protocol Enabling Full-Length Total RNA Sequencing From Single Cells. Front Genet 12, 665888 (2021)
- Epiclomal: Probabilistic clustering of sparse single-cell DNA methylation data. PLoS Comput Biol 16, e1008270 (2020)
- Clonal Decomposition and DNA Replication States Defined by Scaled Single-Cell Genome Sequencing. Cell 179, 1207-1221.e22 (2019)
- Scalable whole-genome single-cell library preparation without preamplification. Nat Methods 14, 167-173 (2017)
Mutational Signatures in DNA Repair Deficient cancers
Utilising whole tissue and our recently developed single cell genome sequencing methods, we are interested in the underlying mechanisms of copy number-structural instability in cancer. We are interested in how to optimize the computational discovery of genome-wide structural and point mutational signatures and how signatures can identify treatment opportunities for ovarian and breast cancers. This work is being carried out at bulk and single cell resolution. We are seeking to define the role of instability in modifying local immune responses. The ultimate goal is to identify new ways of targeting the unique biology associated in breast and ovarian cancers.
In our recent work, by sequencing the genomes of individual cancer cells, we identified distinct ‘foreground’ mutational patterns defined by cell-to-cell structural variation occurring in the ‘background’ of cancer-associated genomic instability. These patterns were found to significantly contribute to the origins of phenotypic and evolutionary diversity of TNBC and HGSC, revealing previously hidden genomic states of cancer cells.
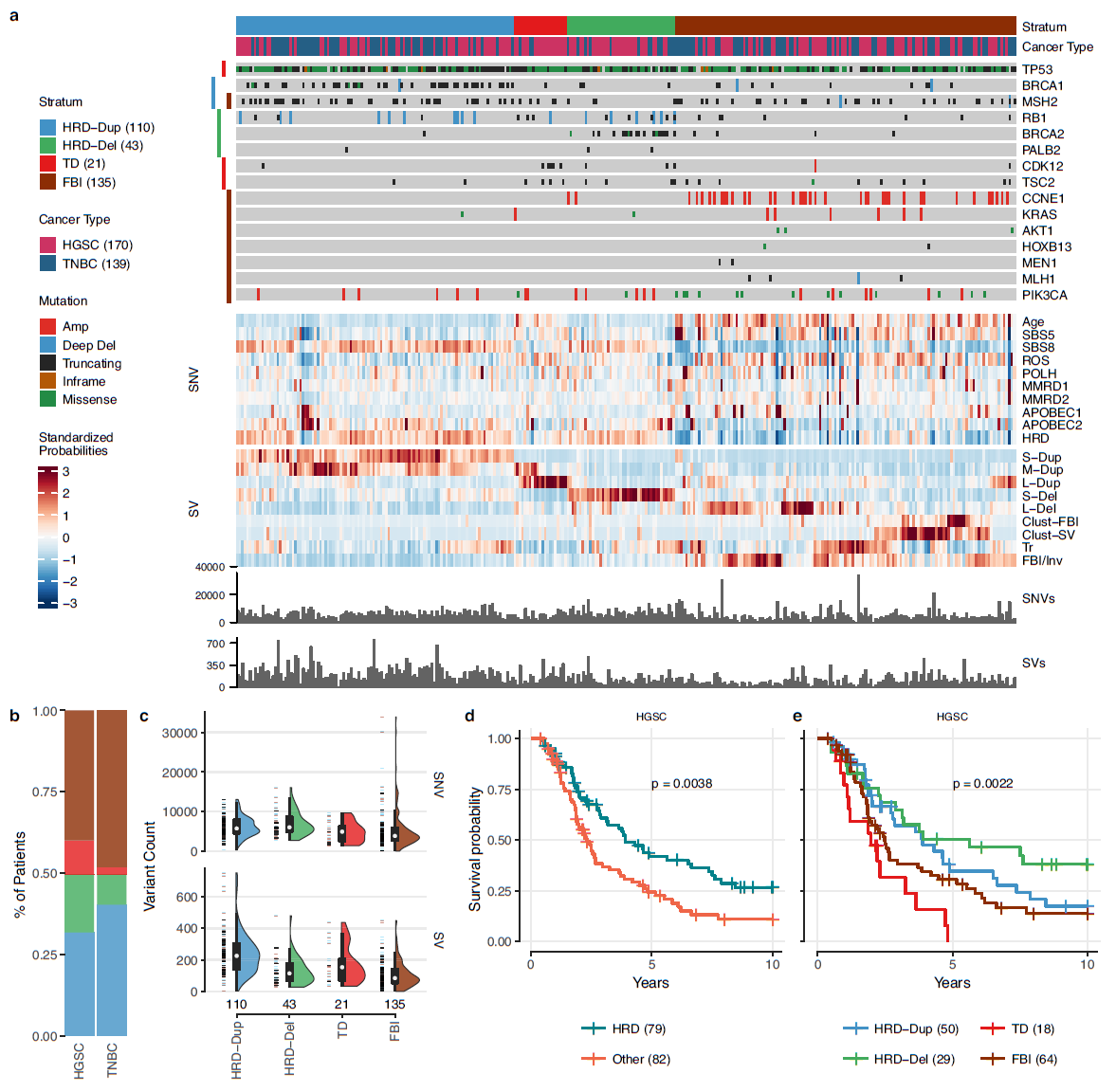
From Funnell et al. (2022, Nature), meta-cohort signature analysis of 139 TNBC and 170 HGSC bulk whole genomes, stratified into different mutational signatures predictive of survival outcomes.
Relevant recent work:
- Single-cell genomic variation induced by mutational processes in cancer. Nature 612, 106-115 (2022)
- Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat Genet 49, 856-865 (2017)
Therapeutic Targeting of Unstable Genomes with G-quadruplex Stabilizers
Using a combination of genetic screening approaches and recent insights into direct targeting of tertiary G-quadruplex structures in the genome, we are identifying new ways to help patients whose tumours do not respond or have become resistant to standard of care therapies. Our work led us to identify G-quadruplex binding small molecule drugs as synthetic lethal to tumours with loss of genome maintenance and repair functions, including homologous recombination, NHEJ, theta-mediated end joining. With support from StandUp to Cancer, we conducted a phase 1 clinical study that provided evidence of mechanism and activity. We also identified splicing reaction modulators with potential in unstable cancers, including novel small molecule inhibitors of CDK12 and CLK2. These approaches are being extended with improved small molecule development and into early stage translation.
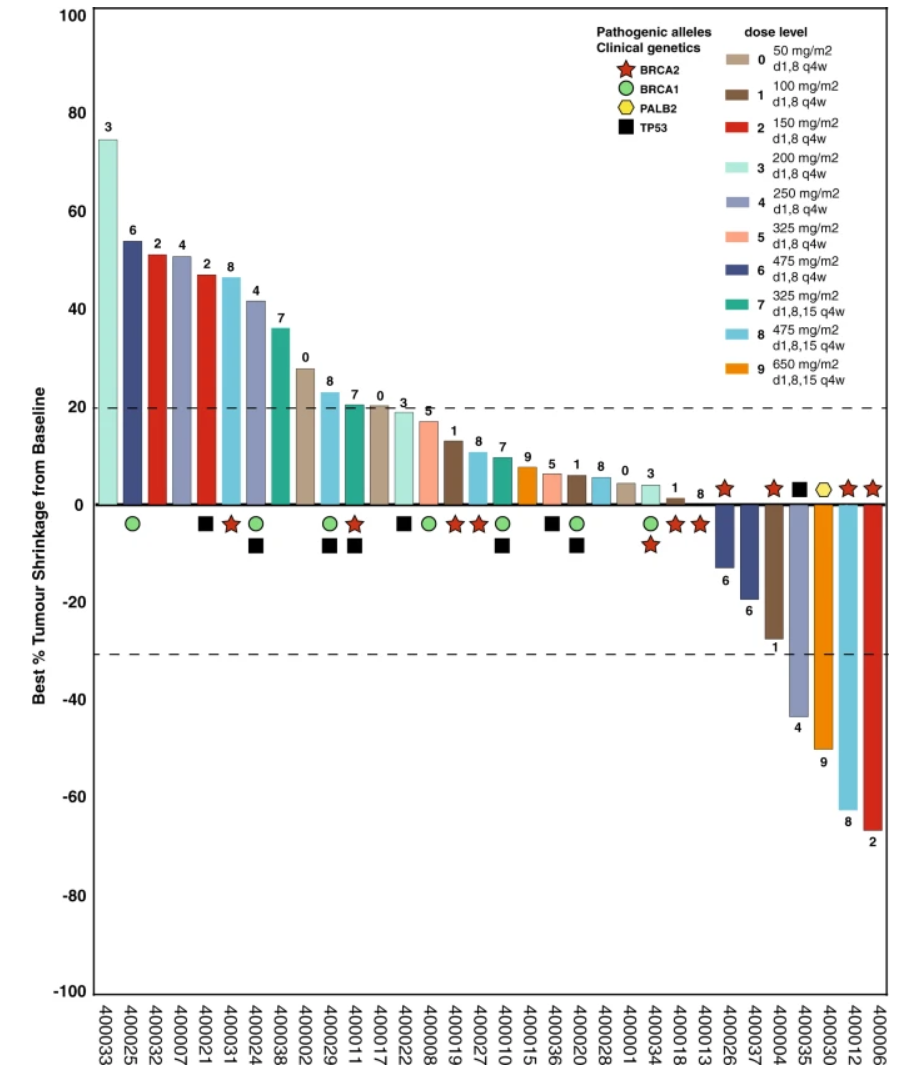
From Hilton et al. (2022), our CX-5461 Stage 1 Clinical Trial shows best responses for the initial cohort, showing CX-5461 effectively predominantly in patients with DNA deficiency repair mutations.
Relevant recent work:
- Results of the phase I CCTG IND.231 trial of CX-5461 in patients with advanced solid tumors enriched for DNA-repair deficiencies. Nature communications 13, 3607 (2022)
- Ubiquitin-mediated DNA damage response is synthetic lethal with G-quadruplex stabilizer CX-5461. Sci Rep 11, 9812 (2021)
- CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun 8, 14432 (2017)
Key collaborators
Vancouver: David Huntsman Poul Sorensen Carl Hansen Alexandre Cote-Bouchard Marco Marra Peter Stirling Mike Underhill Martin Hirst Andy Roth Karen Cheung Phil Hieter
Canada: Dave Cescon Morag Park Michael Pollak Lincoln Stein Anne-Marie Mes-Masson
International: Greg Hannon Carlos Caldas Sohrab Shah Joan Brugge Shankar Balasubramanian Charlie Swanton Simon Tavare Bernd Bodenmiller Johanna Joyce Ed Boyden Xiaowei Zhuang Phil Quirke
Funding sources


