Abstract
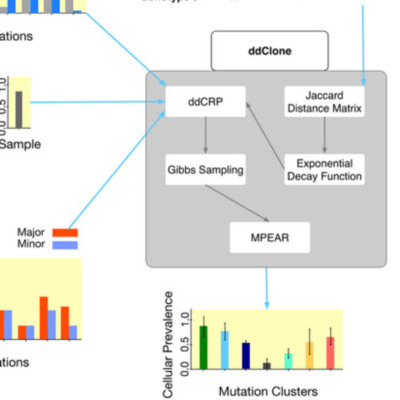
Next-generation sequencing (NGS) of bulk tumour tissue can identify constituent cell populations in cancers and measure their abundance. This requires computational deconvolution of allelic counts from somatic mutations, which may be incapable of fully resolving the underlying population structure. Single cell sequencing (SCS) is a more direct method, although its replacement of NGS is impeded by technical noise and sampling limitations. We propose ddClone, which analytically integrates NGS and SCS data, leveraging their complementary attributes through joint statistical inference. We show on real and simulated datasets that ddClone produces more accurate results than can be achieved by either method alone.