Abstract
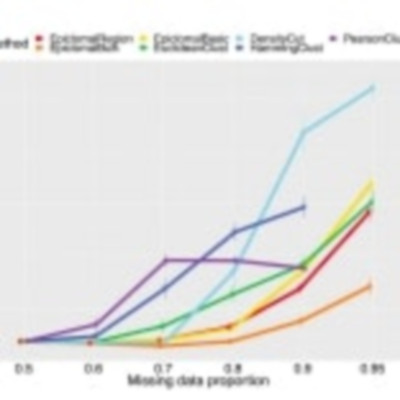
We present Epiclomal, a probabilistic clustering method arising from a hierarchical mixture model to simultaneously cluster sparse single-cell DNA methylation data and impute missing values. Using synthetic and published single-cell CpG datasets, we show that Epiclomal outperforms non-probabilistic methods and can handle the inherent missing data characteristic that dominates single-cell CpG genome sequences. Using newly generated single-cell 5mCpG sequencing data, we show that Epiclomal discovers sub-clonal methylation patterns in aneuploid tumour genomes, thus defining epiclones that can match or transcend copy number-determined clonal lineages and opening up an important form of clonal analysis in cancer. Epiclomal is written in R and Python and is available at https://github.com/shahcompbio/Epiclomal.