Abstract
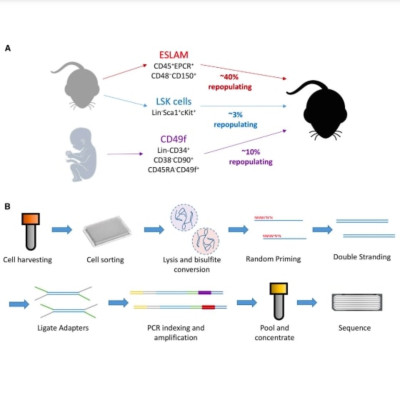
Increasing evidence of functional and transcriptional heterogeneity in phenotypically similar cells examined individually has prompted interest in obtaining parallel methylome data. We describe the development and application of such a protocol to index-sorted murine and human hematopoietic cells that are highly enriched in their content of functionally defined stem cells. Utilizing an optimized single-cell bisulfite sequencing protocol, we obtained quantitative DNA methylation measurements of up to 5.7 million CpGs in single hematopoietic cells. In parallel, we developed an analytical strategy (PDclust) to define single-cell DNA methylation states through pairwise comparisons of single-CpG methylation measurements. PDclust revealed that a single-cell epigenetic state can be described by a small (<1%) stochastically sampled fraction of CpGs and that these states are reflective of cell identity and state. Using relationships revealed by PDclust, we derive near complete methylomes for epigenetically distinct subpopulations of hematopoietic cells enriched for functional stem cell content.