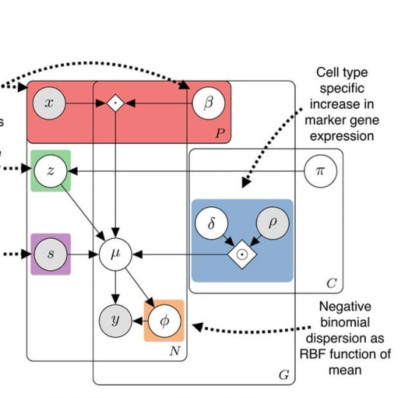
Abstract
Single-cell RNA sequencing has enabled the decomposition of complex tissues into functionally distinct cell types. Often, investigators wish to assign cells to cell types through unsupervised clustering followed by manual annotation or via ‘mapping’ to existing data. However, manual interpretation scales poorly to large datasets, mapping approaches require purified or pre-annotated data and both are prone to batch effects. To overcome these issues, we present CellAssign, a probabilistic model that leverages prior knowledge of cell-type marker genes to annotate single-cell RNA sequencing data into predefined or de novo cell types. CellAssign automates the process of assigning cells in a highly scalable manner across large datasets while controlling for batch and sample effects. We demonstrate the advantages of CellAssign through extensive simulations and analysis of tumor microenvironment composition in high-grade serous ovarian cancer and follicular lymphoma.