Abstract
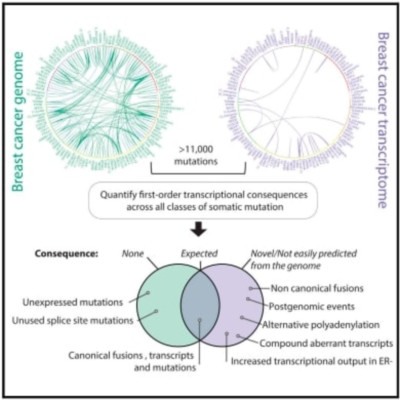
Disordered transcriptomes of cancer encompass direct effects of somatic mutation on transcription, coordinated secondary pathway alterations, and increased transcriptional noise. To catalog the rules governing how somatic mutation exerts direct transcriptional effects, we developed an exhaustive pipeline for analyzing RNA sequencing data, which we integrated with whole genomes from 23 breast cancers. Using X-inactivation analyses, we found that cancer cells are more transcriptionally active than intermixed stromal cells. This is especially true in estrogen receptor (ER)-negative tumors. Overall, 59% of substitutions were expressed. Nonsense mutations showed lower expression levels than expected, with patterns characteristic of nonsense-mediated decay. 14% of 4,234 rearrangements caused transcriptional abnormalities, including exon skips, exon reusage, fusions, and premature polyadenylation. We found productive, stable transcription from sense-to-antisense gene fusions and gene-to-intergenic rearrangements, suggesting that these mutation classes drive more transcriptional disruption than previously suspected. Systematic integration of transcriptome with genome data reveals the rules by which transcriptional machinery interprets somatic mutation.