Abstract
High mitotic activity is associated with the genesis and progression of many cancers. Small molecule inhibitors of mitotic apparatus proteins are now being developed and evaluated clinically as anticancer agents. With clinical trials of several of these experimental compounds underway, it is important to understand the molecular mechanisms that determine high mitotic activity, identify tumor subtypes that carry molecular aberrations that confer high mitotic activity, and to develop molecular markers that distinguish which tumors will be most responsive to mitotic apparatus inhibitors.
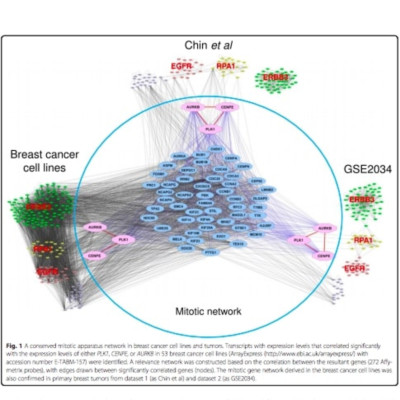
We identified a coordinately regulated mitotic apparatus network by analyzing gene expression profiles for 53 malignant and non-malignant human breast cancer cell lines and two separate primary breast tumor datasets. We defined the mitotic network activity index (MNAI) as the sum of the transcriptional levels of the 54 coordinately regulated mitotic apparatus genes. The effect of those genes on cell growth was evaluated by small interfering RNA (siRNA).
High MNAI was enriched in basal-like breast tumors and was associated with reduced survival duration and preferential sensitivity to inhibitors of the mitotic apparatus proteins, polo-like kinase, centromere associated protein E and aurora kinase designated GSK462364, GSK923295 and GSK1070916, respectively. Co-amplification of regions of chromosomes 8q24, 10p15-p12, 12p13, and 17q24-q25 was associated with the transcriptional upregulation of this network of 54 mitotic apparatus genes, and we identify transcription factors that localize to these regions and putatively regulate mitotic activity. Knockdown of the mitotic network by siRNA identified 22 genes that might be considered as additional therapeutic targets for this clinically relevant patient subgroup.
We define a molecular signature which may guide therapeutic approaches for tumors with high mitotic network activity.