Abstract
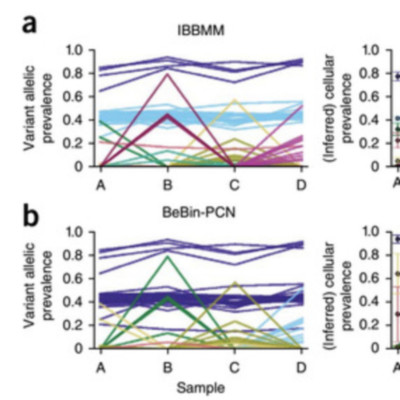
We introduce PyClone, a statistical model for inference of clonal population structures in cancers. PyClone is a Bayesian clustering method for grouping sets of deeply sequenced somatic mutations into putative clonal clusters while estimating their cellular prevalences and accounting for allelic imbalances introduced by segmental copy-number changes and normal-cell contamination. Single-cell sequencing validation demonstrates PyClone’s accuracy.