Abstract
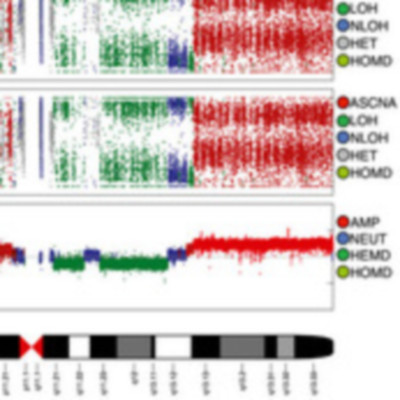
Loss of heterozygosity (LOH) and copy number alteration (CNA) feature prominently in the somatic genomic landscape of tumors. As such, karyotypic aberrations in cancer genomes have been studied extensively to discover novel oncogenes and tumor-suppressor genes. Advances in sequencing technology have enabled the cost-effective detection of tumor genome and transcriptome mutation events at single-base-pair resolution; however, computational methods for predicting segmental regions of LOH in this context are not yet fully explored. Consequently, whole transcriptome, nucleotide-level resolution analysis of monoallelic expression patterns associated with LOH has not yet been undertaken in cancer. We developed a novel approach for inference of LOH from paired tumor/normal sequence data and applied it to a cohort of 23 triple-negative breast cancer (TNBC) genomes. Following extensive benchmarking experiments, we describe the nucleotide-resolution landscape of LOH in TNBC and assess the consequent effect of LOH on the transcriptomes of these tumors using RNA-seq-derived measurements of allele-specific expression. We show that the majority of monoallelic expression in the transcriptomes of triple-negative breast cancer can be explained by genomic regions of LOH and establish an upper bound for monoallelic expression that may be explained by other tumor-specific modifications such as epigenetics or mutations. Monoallelically expressed genes associated with LOH reveal that cell cycle, homologous recombination and actin-cytoskeletal functions are putatively disrupted by LOH in TNBC. Finally, we show how inference of LOH can be used to interpret allele frequencies of somatic mutations and postulate on temporal ordering of mutations in the evolutionary history of these tumors.