Abstract
Identification of somatic single nucleotide variants (SNVs) in tumour genomes is a necessary step in defining the mutational landscapes of cancers. Experimental designs for genome-wide ascertainment of somatic mutations now routinely include next-generation sequencing (NGS) of tumour DNA and matched constitutional DNA from the same individual. This allows investigators to control for germline polymorphisms and distinguish somatic mutations that are unique to the tumour, thus reducing the burden of labour-intensive and expensive downstream experiments needed to verify initial predictions. In order to make full use of such paired datasets, computational tools for simultaneous analysis of tumour-normal paired sequence data are required, but are currently under-developed and under-represented in the bioinformatics literature.
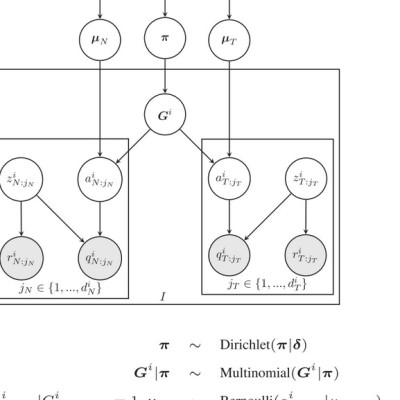
In this contribution, we introduce two novel probabilistic graphical models called JointSNVMix1 and JointSNVMix2 for jointly analysing paired tumour-normal digital allelic count data from NGS experiments. In contrast to independent analysis of the tumour and normal data, our method allows statistical strength to be borrowed across the samples and therefore amplifies the statistical power to identify and distinguish both germline and somatic events in a unified probabilistic framework.
The JointSNVMix models and four other models discussed in the article are part of the JointSNVMix software package available for download at http://compbio.bccrc.ca
sshah@bccrc.ca
Supplementary data are available at Bioinformatics online.