Abstract
The study of cancer genomes now routinely involves using next-generation sequencing technology (NGS) to profile tumours for single nucleotide variant (SNV) somatic mutations. However, surprisingly few published bioinformatics methods exist for the specific purpose of identifying somatic mutations from NGS data and existing tools are often inaccurate, yielding intolerably high false prediction rates. As such, the computational problem of accurately inferring somatic mutations from paired tumour/normal NGS data remains an unsolved challenge.
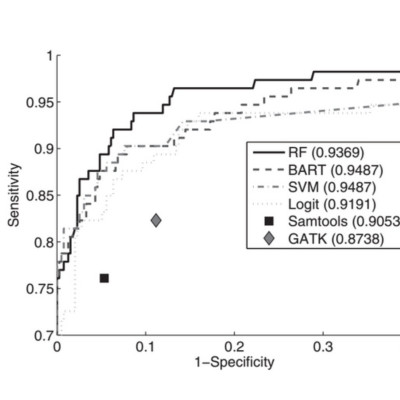
We present the comparison of four standard supervised machine learning algorithms for the purpose of somatic SNV prediction in tumour/normal NGS experiments. To evaluate these approaches (random forest, Bayesian additive regression tree, support vector machine and logistic regression), we constructed 106 features representing 3369 candidate somatic SNVs from 48 breast cancer genomes, originally predicted with naive methods and subsequently revalidated to establish ground truth labels. We trained the classifiers on this data (consisting of 1015 true somatic mutations and 2354 non-somatic mutation positions) and conducted a rigorous evaluation of these methods using a cross-validation framework and hold-out test NGS data from both exome capture and whole genome shotgun platforms. All learning algorithms employing predictive discriminative approaches with feature selection improved the predictive accuracy over standard approaches by statistically significant margins. In addition, using unsupervised clustering of the ground truth ‘false positive’ predictions, we noted several distinct classes and present evidence suggesting non-overlapping sources of technical artefacts illuminating important directions for future study.
Software called MutationSeq and datasets are available from http://compbio.bccrc.ca.