Abstract
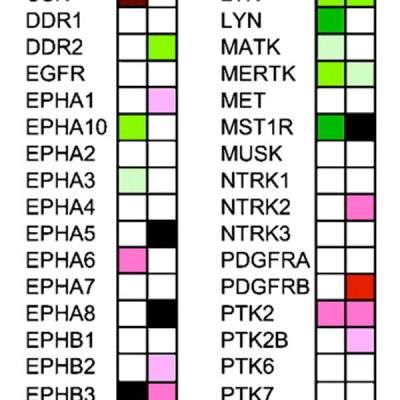
The insulin-like growth factor-1 receptor (IGF1R) is emerging as a promising therapeutic target in human cancers. In the high-risk childhood sarcomas Ewing family tumor and rhabdomyosarcoma, IGF1R-blocking antibodies show impressive antitumor activity in some but not all patients, and acquired resistance is observed. Because tumor IGF1R mutations are not described, the basis of IGF1R inhibitor resistance remains unknown. We hypothesized that compensatory signaling cascades bypassing targeted IGF1R inhibition might be involved. To test this systematically, we performed small interfering RNA (siRNA) screens in sarcoma cell lines to identify IGF1R pathway components or related protein tyrosine kinase (PTK) networks that modulate the antitumor efficacy of the BMS-536924 IGF1R kinase inhibitor. This strategy revealed (a) that sarcoma cells are exquisitely sensitive to loss of distal rather than proximal IGF1R signaling components, such as ribosomal protein S6 (RPS6); (b) that BMS-536924 fails to block RPS6 activation in resistant sarcoma cell lines; and (c) that siRNA knockdown of the macrophage-stimulating 1 receptor tyrosine kinase (MST1R; also known as RON) restores BMS-536924 efficacy, even in highly drug-resistant cell lines. We confirmed MST1R expression across a broad panel of childhood sarcomas, and found that loss of MST1R by RNA interference blocks downstream RPS6 activation when combined with BMS-536924 in vitro. These findings underscore the importance of fully understanding PTK networks for successful clinical implementation of kinase inhibitor strategies.